
wildscienceblog
Wildscience blog by Tania Guerrero
Hi there! I am still using Tumblr in plain 2020 as a blog but soon will have my onw website just like cool scientists do (except I'm missing the cool and in the view of some also the scientist part, but who cares?). Anyway, I am Tania born and raised in good and crazy Mexico City (actually mostly in the State of Mexico but only locals get how that works), I studied Veterinary Medicine but make no mistake, I am a nerd rather than a clinician. I've always been interested in the interplay between genetic makeup and environmental cues to understand health and survival outcomes in both wildlife and domesticated populations. In 2019, I obtained my MSc in Environmental Sciences working on cheetah epigenetics! I am currently particularly keen on the molecular mechanisms regulating and linking this interplay, its transgenerational effects and consequences, and in mismatches between earlier adaptations and the present environment. I used to use this website to post fun and curious facts or news about animals with a sprinkle of molecular biology but expect mainly amateur content as my post are rather old.
211 posts
Don't wanna be here? Send us removal request.
Last Seen Blogs

f-ai-n
Fain is one letter away from Pain

alannadale
I'm Your Lionheart

bratzhotwife
🪷Brat_z_Hotwife🪷

allanrandall-percussion
Allan Randall- Percussion

babywinona
Winona Ryder Defense Squad
Text
My unpopular opinion about popular science topics: A new study says that SARS-CoV-2 RNA may integrate into the human genome - DON’T PANIC, this is likely not the case and even if it was, would probably lack clinical relevance :) -
For a few days now, there has been an ongoing heated discussion on Academic Twitter and other scientific outreach outlets regarding a bioRxiv preprint titled “SARS-CoV-2 RNA reverse-transcribed and integrated into the human genome” (Zhang et al. bioRxiv, 2020). The study proposes that the sustained viral RNA shedding and recurrence of PCR positive testing after recovery might be due to retrointegration (in simple terms this is the insertion of DNA sequences into a host genome mediated by an RNA intermediate, the enzymes reverse-transcriptase and endonuclease and chaperone proteins) of SARS-CoV-2 into the DNA of patients (i.e. their nuclear DNA). The senior author is the renowned scientist Rudolf Jaenisch who together with Beatrice Mintz, produced the first-ever transgenic mammals by injecting retroviral DNA into early mice embryos. He is a professor of biology at MIT and his lab based in the Whitehead Institute for Biomedical Research works on epigenetics and epigenome editing and transgenic models to understand neurological disorders and cancer and has recently included research on SARS-CoV-2. This research focus is shared by his co-authors, who also are postdoctoral researchers at his lab. The first author, Liguo Zhang studies epigenetics and nuclear organization in the nervous system and how their misregulation leads to diseases, followed by Alexsia Richards who is currently examining the tropism and transcriptional response to SARS-CoV-2, Andrew Khalil focuses on understanding the role of the adaptive immune system in regulating metabolism, Emile Wogram combines stem cell technologies, genome engineering, biochemistry, and proteomics to study microglial phagocytosis, and Haiting Ma is concerned with studying signaling pathways and epigenetics of stems cells differentiation and maturation into functional lineages. Richard A. Young also a professor of biology at MIT and a member of the Whitehouse Institute pioneer in the systems biology of gene control in health and disease also participated in this research.
Nevertheless, these findings are raising many eyebrows among the scientific community, especially under the current climate where an mRNA-based vaccine against SARS-CoV-2 is already approved in some countries and another one to be soon authorized (see here and here if you want to learn more of the current status on vaccines’ roll out). Scientists from fields ranging from immunology to cancer biology to the closest it gets to the topics being addressed in the polemic study, mobile elements, transposons, and endogenous viruses as well as clinical virology, have sharply criticized the soundness of their conclusions and are calling for the retraction of the preprint.
A major concern is the adequacy of the experimental assays used to support the hypothesis of retrotransposition events. Zhang et al. (bioRxiv, 2020) report to have found chimeric transcripts of fused viral and cellular sequences in published data of cultured cells and primary cells of patients. They overexpressed human LINE-1 or HIV-1 reverse transcriptase in cells which were then infected with SARS-CoV-2 and applied a single-molecule RNA-FISH (i.e. an in situ hybridization method that uses probes of multiple oligonucleotides to detect individual RNA molecules inside of single cells https://sites.google.com/site/singlemoleculernafish/home?authuser=0) to confirm that viral sequences were integrated and detected their transcription in the nucleus of the cells overexpressing LINE-1. They also analyzed published data on LINE-1 expression in cells infected with SARS-CoV-2 and chimeric read abundance and found a correlation between these two. Furthermore, they suggest a molecular mechanism by which LINE-1 expression can be stimulated under SARS-CoV-2 infection via cytokines. However, Cedric Feschotte a professor at the Department of Molecular Biology and Genetics in Cornell University with 20 years of experience studying mobile elements points out that these experimental approaches are insufficient to sustain an in vivo retrotransposition because they omit gold standard techniques such as isolating and reporting of “the sequence of the integrants along with flanking genomic regions (junction sequences spanning both viral and flanking DNA)” and fail to present hallmarks of the proposed LINE-1 dependent process (e.g. short direct repeats flanking integrant, integration at preferred L1 endonuclease cleavage site [TTTT/AA], polyA tail at the 3’ end or chimera with the 3’ end of endogenous L1/Alu). A generalized opinion among critics of Zhang et al. (bioRxiv, 2020) is that the chimeric reads are likely to be artifacts of library preparation or template-switching events and that their approach does not rule out these possibilities.
Aris Katzourakis, a professor of Evolution and Genomics at the University of Oxford whose research is centered in the study of ancient viruses and uses endogenous viral elements, including retroviruses tweeted: “ … genomic integration of coronaviral DNA is highly implausible, given there is not a single known genomic fossil of integrated coronaviruses in any know host genome that has ever been sequenced to date.” Which is a strong and educated argument (yet not a smoking gun) against the evidence presented by Zhang et al. (bioRxiv, 2020). Moreover, various academics argue that even if substantiated under experimental conditions, is a whole different thing to prove retrotransposition happening during natural infection with SARS-CoV-2. Clinical scientists draw attention that is definitely not unique to SARS-CoV-2 the persistency of viral debris in tissues of convalescent or recovered patients (even though its mechanism and clinical relevance remain subject to discussion e.g. see here). Altogether, they agree that its clinical relevance is far from proven and unlikely to exist.
As dull as it is for me, I am merely a spectator here since, despite my keen curiosity for genetic mobile elements, I have very limited first-hand experience on these subjects. I do think that a take-home message is that probably many features described as novel or unique to SARS-CoV-2 (which sometimes alarm scientists and the general public alike) might be in fact more ubiquitous than the flood of SARS-CoV-2 scientific papers would have us believe. In my opinion, the reason being that the acuteness and the timing in terms of technology and communications of this pandemic have powered studies at scales that have not been possible to conduct in other disease outbreaks. In other words, we are looking too much, too close, and perhaps missing a bigger picture (I have seen a strikingly low number of papers building upon previous knowledge on other coronaviruses, especially at the clinical level). Whether or not SARS-CoV-2 integrates into our genomes and if so, whether this has clinical implications, is definitely something that cannot yet and should not be answered based on the findings by Zhang et al. (bioRxiv, 2020). It is also safe to say, that whereas the semantics can be confusing for the general public, even if retrotransposition is later proven to occur in vivo during natural infections, there is still a long shot from there to assert that this implies a direct risk for mRNA vaccines to integrate into our genomes because they are designed such that they do not interact with our DNA (they do not go inside the nucleus, you can read more about the different vaccines and their mechanisms here). Having said that, clinical usage of mRNA vaccines is new, and we will be confirming and learning more about how they (hopefully well!) work soon.
Lastly, I want to share that my motivation to write this piece was not so much the hot topic that SARS-CoV-2 is but rather the atmosphere that this paper as others in various fields which present (seemingly) premature conclusions and bold takes have triggered. This paper has the feature of being presented as a preprint. Preprints have the goal of allowing the scientific audience “to see, discuss, and comment on the findings immediately” before peer-review is completed. Therefore, while I find most critiques to this paper very compelling and I strongly agree that when a bias or unsubstantiated evidence is identified, this should be acknowledged and corrected, I consider a poor scientific practice to demand a retraction of a publication at a stage that has precisely the aim of calling for discussion. I am talking now of a number of papers I have seen being targeted by similar reactions. The intentions of authors cannot be, of course, inferred, so why not waiting for them to take part in the discussion before deploying what seems like attempts to bowdlerize rather than enriching, correcting, or helping improve. Has not this been the case of most wondrous and prodigious scientific findings and technological advances? In the past, findings that were about to change the world but too odd for their surrounding context (please let me be clear that I am not referring to any particular paper that I believe to have such potential, but rather to a general attitude towards outliers of the mainstream) were met by generalized disbelief and mockery. I thought that we have already moved away from the practices that discourage sharing surprising results. Of course, there is a thin line between sharing to prompt healthy discussions and incurring unethical practices, but where are going to end up if we try to ban everything that does not comply with our preconceptions? And even if claims end up being wrong, how are we supposed to ensure that they are corrected if we promote distrust and an unwelcoming arena. Is this a retrogression powered by a more than ever intolerant society? or it never left and is it just becoming evident as communications allow it? I think we should do better.
2 notes
·
View notes
Photo

That’s how my news feed on Facebook and Twitter looks like, pretty cool findings both :) I leave you here the links to the press releases and original papers.
New genus of Australian lion:
A little spoiler: just don’t get too excited about the Australian lion, it is unfortunately not an extant species. It was a little fella, one of the tiniest marsupial lions from the Oligocene-Miocene with a presumable omnivory tendency :)
https://www.abc.net.au/news/2020-02-28/australian-lion-discovered-in-outback-queensland/12007324
https://www.tandfonline.com/doi/abs/10.1080/02724634.2019.1703722
Red pandas:
Before being placed in a family on their own (Ailuridae), Red pandas wandered around the phylogenetic tree for some time. They have been classified as close relatives to the Giant panda (among other reasons, because they both have a “false thumb” and feed on bamboo) and to the raccoon,...yeah they look kinda similar and farther back in time (around 33 million years ago) they might share some common ancestors.
https://www.bbc.com/news/science-environment-51632790
https://advances.sciencemag.org/content/6/9/eaax5751
And now a picture of two cute red panda cubs:

0 notes
Link
Scientists have created a living organism whose DNA is entirely human-made — perhaps a new form of life, experts said, and a milestone in the field of synthetic biology.
Researchers at the Medical Research Council Laboratory of Molecular Biology in Britain reported on Wednesday that they had rewritten the DNA of the bacteria Escherichia coli, fashioning a synthetic genome four times larger and far more complex than any previously created.
The bacteria are alive, though unusually shaped and reproducing slowly. But their cells operate according to a new set of biological rules, producing familiar proteins with a reconstructed genetic code.
Continue Reading.
2K notes
·
View notes
Photo

His name is Bond, Plagiodontia aedium bondi
Meet the cutest James Bond of all times! Ok, this James Bond may not drive extra luxury and equipped cars nor save the world without a scratch to his tuxedo, but this one, is definitely the most charming.
Plagiodontia aediumbondi is a cat-sized rodent newly discovered on the Caribbean island of Hispaniola and as it turns out it both this James Bond and the fictional spy from the movies owe their names to a real-life naturalist.
Samuel Turvey of the Zoological Society of London and his team described this species in the Journal Zootaxta, which weighs more than a kilogram, is a type of hutia, a family of secretive and sadly scarce rodents that live mainly in the West Indian islands.
There used to be more than 30 species, however nowadays most hutia have been driven to extinction by the colonization of these islands. The newly discovered resident may be one of only eight types of hutia left.
“I am glad we were able to describe James Bond’s hutia before it’s too late, as it is highly threatened by deforestation, even in protected areas,” says Turvey.

Reference article here
3 notes
·
View notes
Photo





The raven is sometimes known as “the wolf-bird.” Ravens, like many other animals, scavenge at wolf kills, but there’s more to it than that.
Both wolves and ravens have the ability to form social attachments and they seem to have evolved over many years to form these attachments with each other, to both species’ benefit.
There are a couple of theories as to why wolves and ravens end up at the same carcasses. One is that because ravens can fly, they are better at finding carcasses than wolves are. But they can’t get to the food once they get there, because they can’t open up the carcass. So they’ll make a lot of noise, and then wolves will come and use their sharp teeth and strong jaws to make the food accessible not just to themselves, but also to the ravens.
Ravens have also been observed circling a sick elk or moose and calling out, possibly alerting wolves to an easy kill. The other theory is that ravens respond to the howls of wolves preparing to hunt (and, for that matter, to human hunters shooting guns). They find out where the wolves are going and following. Both theories may be correct.
Wolves and ravens also play. A raven will sneak up behind a wolf and yank its tail and the wolf will play back. Ravens sometimes respond to wolf howls with calls of their own, resulting in a concert of howls and calls.
Sources: Mind of the Raven, Bernd Heinrich, The American Crow and the Common Raven, Lawrence Kilham
66K notes
·
View notes
Photo




Badger cubs have arrived at Wildlife Aid!
We now have four young badger cubs at the centre. The first of these was collected by the RSPCA before being transferred here. It had been found alone in the middle of a farmyard track where, sadly, there had been no signs of an adult.
It had a lot of ticks and fleas, but otherwise was in good shape. He has now been joined by three other cubs from South Essex Wildlife Hospitaland they are making a complete mess of our pen!
You can watch them live (along with our fox cubs and two bird nests) on our online webcams :)
114 notes
·
View notes
Link
0 notes
Text
Capture-Recapture-Mark using DNA genotyping: Challenges and State of the Art
This is one of those topics that I would consider “old but gold”. I recently had to write about it for one of my classes. So I thought sharing the information I compiled with you too :)
One of the most popular techniques when scientists are looking for quantitative (although as we will see, it ends up being semiquantitative or quasi-quantitative due to technical constraints) population studies is Capture-Mark-Recapture which has two main variants: traditional CMR, and Model-Based CMR, commonly used along genotyping. So, I decided to explore further into the later, because I noticed that even though it’s become very common practice among monitoring programs, is still having many shortcomings, so I wanted to understand the source of these and how researchers are coping with them in the recent years.
Genetic Capture-Mark-Recapture (CMR), as said before is now a routine procedure in many programs of wildlife conservation. Some of the most common applications of this technique are: estimates of population size (abundance), sex ratio, survival, migrations patterns, fecundity, population growth and of course the genotypes can be used for paternity tests (Lampa et al., 2015; Lucaks and Burnham., 2005).
The general steps of the method imply processing non-invasively collected samples (e.g. faeces, hair, feathers, saliva, etc.) to extract DNA and genotype it at multiple loci (e.g. microsatellites). This multilocus genotype is then considered a molecular individual mark, just as fingerprints in forensic medicine. After obtaining a number of samples, matching genotypes are considered to belong to the same individual and are classified as recaptures. While non-matching genotypes should indicate newly captured animals, but unfortunately that is not always as straight forward.
So, even nowadays, there are still challenges to overcome when using genotyping-CMR. Genotyping errors can be misleading, causing to erroneously assign a sample to the wrong individual. Also can create “false individuals” by just a single loci being mistyped.
Samples can also be confounded if they contain a mixture of DNA from more than one individual. It can also be an intrinsic bias, as in human forensics some individuals had been identified and termed “shedders” which tend to leave more DNA traces than others (“nonshedders”) (Lucaks and Burnham, 2005).
The problems faced in the genetic analysis itself, of noninvasive samples can arise from very different sources and steps. Some of the most commonly referenced are: amplification failure and allelic dropout (Lukacs and Burnham, 2005). Amplification failure can be caused by a lack of the target sequence (due to degraded DNA) or by PCR inhibitors present in the sample. While allelic dropout, is by definition the loss of one allele during polymerase chain reaction (PCR) amplification of DNA (Steyer, K, et al. 2016).
Challenges in constructing capturing histories from multilocus genotypes may also involve inflated mark-recapture abundance estimates. Additionally, while increasing the number of analyzed loci improves the discriminating power of the test, larger panels also increase the probability of additional genotyping error events (Sethi, et al. 2016).
Possible solutions to overcome genotyping errors
Approaches to deal with genotyping most common sources of error have been proposed, which can be grouped into the following categories: (i) removing genotyping errors from the data, (ii) develop robust genotyping errors sampling protocols, but this is not as straight forward as it seems (Lampa et al., 2015; Sethi, 2016).
There are some tests that can provide useful information about the quality of the genotypes. One of these is the examining-bimodality (EB) test, that searches for an over-abundance of genotypes observed only once. Second one is the Difference in Capture History (DCH) which examines the rate at which new individuals are recognized by looking at more loci (McKelvey and Schwartz, 2004). Although the two tests assume equal capture probability among individuals (which seems to be one of the assumptions that researchers can’t shake off when using CMR).
Since the type of errors mainly depend on the stage of the process we look at, (i.e. sampling, genotyping and in most cases, estimating population size) it makes sense to approach the solutions on the same basis. For instance, when collecting the DNA samples it is very important to chose a sampling design appropriate for the ecology and other details of target species and the assumptions of the selected CMR model (e.g. closed populations, capture probabilities, etc.). As simple as this step may seem, neglecting it would lead to unsuccessful downstream genotyping and population analysis. Other relevant considerations on early stages are the careful sample handling, preservation and extraction methods, given the fact that samples normally contain contaminants, PCR inhibitors and poor quality genetic material (Lampa et al., 2013).
As seen overestimation or underestimation of populations size are very sensitive to even slight genotyping errors. One recommendation is to look for the best balance of the number of typed loci to minimize the probability of identity without increasing too much the genotyping effort and thus, the error window (Lampa et al., 2013; McKelvey and Schwartz, 2004).
There are attempts harnessing bioinformatic tools for simulations and exploring options to select the number of markers, allelic richness and genotyping error rates which are acceptable for efficient CMR histories. They tested error-tolerant likelihood-based match, combining probabilities of latent genotypes and probabilities of observed genotypes, which may contain genotyping errors. Since they compared microsatellite based makers and Single-Nucleotide Polymorphisms (SNPs, which are another option for genotyping not so commonly explored in CMR), they found for instance that SNPs are computationally more efficient during the match calling and sample clustering. They suggest that SNPs may overtake microsatellite approaches in the future (Sethi et al., 2016).
Choosing the best models to fit the data obtained by microsatellite genotyping is also very challenging, given that one must take many assumptions (closed population, equal capture probabilities). The general recommendation is to prefer a model accounting for error. For instance, a recently published paper, combined the non-invasive CMR with Spatially Explicit Capture Recapture approach using wolves feces. They applied a Poisson observation model with a single survey, and the null model M0. To overcome the problem with the assumptions that all individuals are uniformly and independently distributed, they modeled each collection site “centroid” of the cell, as count detectors based on the assumption that the same wolf can be detected at multiple cells during the sampling and more than one individual can be detected in the same detector (López et al., 2018).
Almost all the steps can be analyzed using decision diagrams of assumptions and filling in with the specific features of the study, jus as exemplified by Lampa et al., 2013 and Lampa et al., 2015. A summary of the general workflow as recommended by Lampa et al., 2013 can be found above, although this and other similar setups must only be considered as guidance.
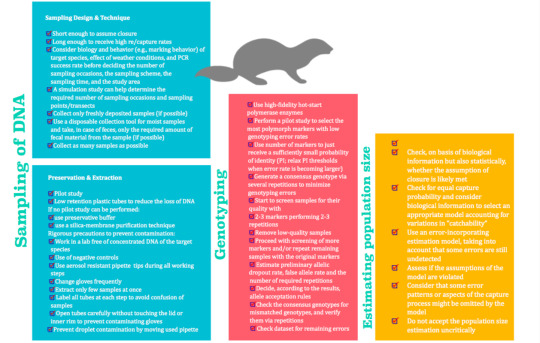
The most recent advances in high-throughput Next Generation Sequencing may help solve some of the genotyping wicked shortcomings by allowing the possibility of sequencing single molecules without previous amplification steps (Lampa, et al., 2013), however I was surprised to found no papers looking specifically into this, so it seems this option still needs to be explored.
In general it seems that is always highly advisable running pilot studies to define the most suitable approach and model prior conducting the actual non-invasive CMR. These pilot studies and good planning overall, can be lifesavers, since it can prevent other sources of bias (inappropriate sample size, wrong type of sample and/or sampling sites, etc.).
As today, although it is very clear that we haven’t reach the point in which genotyped-CMR data can be considered 100 % reliable, as new techniques continue to develop is very likely that in the near future more robust tools will become available.
References
López-Bao JV, Godinho R, Pacheco C, Lema FJ, García E, Llaneza L, Palacios V, Jiménez J (2018), Towards reliable population estimates of wolves by combining spatial capture-recapture models and non-invasive DNA-monitoring. Nature Scientific Reports 8:2177.
Lukacs MP and Burnham PK (2005), Review of capture–recapture methods applicable to noninvasive genetic sampling. Molecular Ecology 14, 3909–3919
Lampa S, Henle K, Klenke R, Hoehn M, Gruber B (2013). How to Overcome Genotyping Errors
in Non-Invasive Genetic Mark-Recapture Population Size Estimation—A Review of Available Methods Illustrated by a Case Study. The Journal of Wildlife Management, DOI: 10.1002/jwmg.604.
Lampa S, Mihoub J-B, Gruber B, Klenke R, Henle K (2015) Non-Invasive Genetic Mark- Recapture as a Means to Study Population Sizes and Marking Behaviour of the Elusive Eurasian Otter (Lutra lutra). PLoS ONE 10(5): e0125684. doi:10.1371/journal.pone.0125684
McKelvey, KS, Schwartz M. K. (2004), Providing Reliable and Accurate Genetic Capture-Mark-Recapture estimates in a cost-effective way. The Journal of Wildlife Management, 68: 453–456. doi 10.2193/0022-541X(2004)068[0453:PRAAGC]2.0.CO;2
Sethi SA, Linden D, Wenburg J, Lewis C, Lemons P, Fuller A, Hare MP (2016) Accurate recapture identification for genetic mark–recapture studies with error-tolerant likelihood-based match calling and sample clustering. R. Soc. open sci. 3: 160457. http://dx.doi.org/10.1098/rsos.160457
Steyer, K., Kraus, R.H.S., Mölich, T. et al. t (2016) Large-scale genetic census of an elusive carnivore, the European wildcat (Felis s. silvestris), Conserv Gene 17: 1183. https://doi.org/10.1007/s10592-016-0853-2
1 note
·
View note
Link
I am sorry my dear DVM colleagues and pro-pure breed supporters, but this is a fact, and I HATE IT!
0 notes
Link
0 notes
Link
Ok... I know this is about humans... but common! it is pretty interesting stuff... so let’s talk a little bit about ancient DNA, shall we? :)
As a logical epidemiological consequence after European contact, several new pathogens were brought to America. To this followed many epidemic outbreaks which had devastating consequences for local, native populations. Studying these diseases and its causal agents pose a great challenge for scientists. On one hand, while some outbreaks were recorded, propose a specific pathogen is very difficult to do when only records of symptoms are available. Adding to this, clinical presentations of certain diseases may have evolved or changed over the years. As a consequence most pathogenic agents that caused these outbreaks still unidentified to date. Thus researchers need to rely on other resources if they are to disentangle these puzzling outbreaks. Here’s where things get interesting because once again DNA tools make their heroic appearance.
Of course the solution didn't come overnight, and first and foremost one needs to have a source of DNA right?.
One of the most interesting cases is the so-called cocoliztli colonial epidemic in Mexico which took place between 1545-1550. This epidemic caused the death of approximately 80% of the native population. Just yesterday Nature Ecology and Evolution published the paper of the international team, led by researchers from the Max Planck Institute for the Science of Human History (MPI-SHH), Harvard University and the Mexican National Institute of Anthropology and History (INAH). They made the most of the only known cemetery linked to this particular outbreak located at Teposcolula-Yucundaa (Oaxaca, México), in order to investigate the pathogen involved in this outbreak.
Samples were obtained from teeth, specifically from the crown pulp chamber. After processing according to DNA archaeological bone protocol and shotgun sequencing the samples, they applied and actually pioneered in a new bioinformatic-metagenomic analysis tool called MALT (Megan ALignment Tool).
The advantage of malt-build (version 0.3.8) is the inclusion of all complete bacterial genomes in the reference database, which in turn allows identifying both pathogenic and non-pathogenic bacteria.
“A key result of this study is that we were successful in recovering information about a microbial infection that was circulating in this population, and we did not need to specify a particular target in advance” explains Alexander Herbig, also of the MPI-SHH and co-first author of the study. In the past, scientists usually targeted a particular pathogen or a small set of pathogens, for which they had a prior indication.
“This new approach allows us to search broadly at the genome level for whatever may be present,” added Johannes Krause, director of the Department of Archaeogenetics at the MPI-SHH and last author of the study. Kirsten Bos, also of the MPI-SHH, adds, “This is a critical advancement in the methods available to us as researchers of ancient diseases – we can now look for the molecular traces of many infectious agents in the archaeological record, which is especially relevant to typical cases where the cause of an illness is not known a priori.”
They went further performing various interesting and very comprehensive methodologies. For example about the phylogeny of Salmonella enterica using 5 ancient genomes. As well as screening for presence/absence of virulence factors using previously identified effector genes. The paper presents genome-wide data from 10 individuals for Salmonella enterica subsp. enterica serovar Paratyphi C, the bacterial cause of the enteric fever. Which they propose as a strong candidate aetiology for the epidemic cocoliztli outbreak at Teposcolula-Yucundaa.
Read the whole paper here
Quotes from authors were obtained from this note from the Max Planck Institute for the Science of Human History here.
Read some background (in Spanish).

Taken from supplementary information Vågene et al, 2018.
0 notes
Link
Enjoy some minutes of relaxation listening to the birds of Mexico City :)
(such a shame that we have so much noise pollution that we can barely hear them...)
0 notes
Quote
It’s hard to know exactly how many axolotls are left in the wild there. Zambrano guesses that during his last survey, in 2014, there were fewer than 1,000 in total, and perhaps fewer than 500. But he can’t be more specific — in the past two years, he’s been unable to raise the money to do any follow-up studies. That he can’t obtain funding for a simple census does not bode well for conservation efforts.
https://www.nature.com/articles/d41586-017-05921-w
How come one of the most widely used lab models, a species that has helped to make countless breakthroughs on human medicine. A species recognized to be undergoing a genetic bottleneck due to inbreeding...DOES NOT GET SUFFICIENT ATTENTION AND FUNDING TO RESTORE AND PROTECT THEIR NATURAL HABITAT?
Someone has to care!!! They deserve better! after all they have done and keep doing for us!
I just wonder… what will all scientists studying (USING) this amazing species, plan on doing when they need toooo...for instance replenishing their genetic pool? (captive bred ones as most lab animals are experiencing inbreeding depression and becoming more susceptible to diseases)…will they care about the natural populations then? or will it be too late?...

Axolotl (Ambystoma mexicanum)
0 notes